Likelihood, Matrix Specification¶
This example use an mxAlgebra likelihood function to compare the fit of two models for the population frequencies of the A, B and O blood groups. It is based on the textbook Likelihood, by Anthony William Fairbank Edwards (1972; 1984). Human beings may be classified according to which of four ABO blood groups they belong: A, B, AB, or O. These groups can be phenotypically distinguished by which antibodies they produce, and this is important for blood transfusions. Patients receiving blood containing an antigen that they do not produce have an adverse reaction to it.
Two hypotheses were advanced to account for the ABO variation. The single locus model posited that there exists one locus with three alleles, A, B and O. Allele A generates antibody A, allele B generates antibody B, and allele O generates neither. Under the two locus model, there is a diallelic A locus, with one allele that produces antibody A, and another that does not. At a second, unlinked, locus B, antigen B is generated by those who have at least one B allele. We try not to make jokes about people who have the BO genotype. For a more thorough account of the ABO blood group system, the Wikipedia article http://en.wikipedia.org/wiki/ABO_blood_group_system is a good starting point.
The two scripts are available in these files:
and
For (former) users of the first version of Mx, scripts for that software can be found here:
Single vs Two Locus Model¶
Data¶
Data from the German mathematician Felix Bernstein <http://en.wikipedia.org/wiki/Felix_Bernstein> (1924) are used for this example. Their observed frequencies are shown in the table, along with expected proportions in the population under the single locus model. A good exercise is to derive the expected proportions by hand. A key consideration is that several different genotypes (combinations of alleles) may generate a single phenotype (observed antigens in the blood). Due to the genetic dominance of allele A, both AA and AO genotypes produce the blood group A phenotype.
Single Locus Model Specification¶
The single locus dominant genetic model, with alleles A and B being codominant (resulting in the AB blood group phenotype) predicts that certain proportions of each blood group will occur in the population. These proportions depend on, and can be expressed as functions of, the allele frequencies. Let the frequencies of alleles A, B and O be 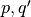 and
and  , respectively. We assume that parents mate at random with respect to their blood groups, so that the likelihood of inheriting two A alleles becomes simply the product of the allele frequencies in the population, i.e.,
, respectively. We assume that parents mate at random with respect to their blood groups, so that the likelihood of inheriting two A alleles becomes simply the product of the allele frequencies in the population, i.e., 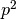 . Thus we are able to draw up a table of the expected proportions of each blood group as a function of the parameters and . These unknown parameters will be estimated from the data using numerical optimization, subject to the constraint that
. Thus we are able to draw up a table of the expected proportions of each blood group as a function of the parameters and . These unknown parameters will be estimated from the data using numerical optimization, subject to the constraint that 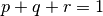 .
.
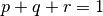
| Phenotype | Genotypes | Frequency | Proportion |
|---|---|---|---|
| A | AO, AA | 234 | p(p+2r) |
| B | BO, BB | 261 | q(q+2r) |
| AB | AB | 182 | 2pq |
| O | OO | 94 | r^2 |
The individuals in this sample are considered to be statistically independent. Therefore the likelihood of observing, e.g., 234 individuals with blood type A, is simply 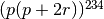 and the log-likelihood is
and the log-likelihood is 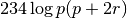 . The OpenMx script will use the four log-likelihoods of the four phenotypes, and sum them to obtain the overall log-likelihood. Optimization proceeds by minimization by default, so we minimize the negative log-likelihood in order to maximize the log-likelihood. The following code block specifies this model. There
. The OpenMx script will use the four log-likelihoods of the four phenotypes, and sum them to obtain the overall log-likelihood. Optimization proceeds by minimization by default, so we minimize the negative log-likelihood in order to maximize the log-likelihood. The following code block specifies this model. There
require(OpenMx)
# Bernstein data on ABO blood-groups
# c.f. Edwards, AWF (1972) Likelihood. Cambridge Univ Press, pp. 39-41
# Matrices for allele frequencies, p, q and r
matP <- mxMatrix( type="Full", nrow=1, ncol=1,
free=TRUE, values=c(.3333), name="P")
matQ <- mxMatrix( type="Full", nrow=1, ncol=1,
free=TRUE, values=c(.3333), name="Q")
matR <- mxMatrix( type="Full", nrow=1, ncol=1,
free=TRUE, values=c(.3333), name="R")
# Matrix of observed data
const <- mxConstraint(P+Q+R == 1, name="EqualityConstraint")
obsFreq <- mxMatrix( type="Full", nrow=4, ncol=1,
values=c(211,104,39,148), name="ObservedFreqs")
# Algebra for predicted proportions
expFreq <- mxAlgebra( expression=rbind(P*(P+2*R), Q*(Q+2*R), 2*P*Q, R*R),
name="ExpectedFreqs")
# Algebra for -2logLikelihood
m2ll <- mxAlgebra( expression=-(sum(log(ExpectedFreqs) * ObservedFreqs)),
name="NegativeLogLikelihood")
# User-defined objective
funAl <- mxFitFunctionAlgebra("NegativeLogLikelihood")
OneLocusModel <- mxModel("OneLocus",
matP, matQ, matR, const, obsFreq, expFreq, m2ll, funAl)
OneLocusFit <- mxRun(OneLocusModel)
OneLocusFit$matrices
OneLocusFit$algebras
Answers should be 0.2945 0.1540 0.5515 for the allele frequencies p, q and r, respectively, and 627.104 for the negative log-likelihood. We now turn to the alternative two-locus model.
Two Locus Model Specification¶
Under the two locus model, we allow for two unlinked (i.e. segregating independently of each other) diallelic loci, A and B. We denote the O allele as a at the A locus, and as b at the B locus, so as to distinguish between these two alleles, neither of which generates an antigen. Thus genotypes at the A locus can be AA, Aa, or aa, with genotype frequencies , 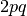 and
and 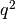 , where
, where 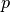 is the proportion of allele in the population, and
is the proportion of allele in the population, and 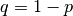 is the proportion of allele a. Similarly, genotypes at the B locus can be BB, Bb or bb, with genotype frequencies
is the proportion of allele a. Similarly, genotypes at the B locus can be BB, Bb or bb, with genotype frequencies 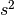 ,
, 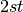 and
and 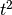 , given allele frequencies
, given allele frequencies  and
and 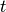 , respectively. Due to the dominance of A over a and B over b, only those with aabb genotypes will belong to blood group O (no antigens). The number the genotype combinations which generate a particular blood group is generally larger than under the single locus model. The combinations, and their expected frequencies in the population, are given in the following table:
, respectively. Due to the dominance of A over a and B over b, only those with aabb genotypes will belong to blood group O (no antigens). The number the genotype combinations which generate a particular blood group is generally larger than under the single locus model. The combinations, and their expected frequencies in the population, are given in the following table:
| Phenotype | Genotypes | Frequency | Proportion |
|---|---|---|---|
| A | AAbb, Aabb | 234 | ( + )*:math:t^2 |
| B | aaBB, aaBb | 261 | 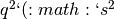 + ) + ) |
| AB | AABB, AABb, AaBB, AaBb | 182 | ( + )( + ) |
| O | aabb | 94 | |
The R script to fit this model is very similar to that of the single locus model. Note, however, that it does not feature the mxConstraint function. There are in fact two constraints, and 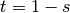 , but these are trivial and easily dealt with using mxAlgebra statements. Although one might think that this approach would be suitable for the single locus model, in which
, but these are trivial and easily dealt with using mxAlgebra statements. Although one might think that this approach would be suitable for the single locus model, in which 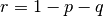 , a difficulty arises because there is no straightforward way to restrict
, a difficulty arises because there is no straightforward way to restrict 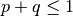 which is necessary for
which is necessary for 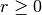 . Models specified so that an allele frequency can go negative during optimization are inherently fragile. A negative allele frequency would potentially result in negative likelihoods, and undefined log-likelihoods. Bounding the parameters to lie between 0.0 and 1.0 provides sufficient robustness to this potential problem.
. Models specified so that an allele frequency can go negative during optimization are inherently fragile. A negative allele frequency would potentially result in negative likelihoods, and undefined log-likelihoods. Bounding the parameters to lie between 0.0 and 1.0 provides sufficient robustness to this potential problem.
require(OpenMx)
# Bernstein data on ABO blood-groups
# c.f. Edwards, AWF (1972) Likelihood. Cambridge Univ Press, pp. 39-41
# Matrices for allele frequencies, p and s
matP <- mxMatrix( type="Full", nrow=1, ncol=1,
free=TRUE, values=c(.3333), name="P")
matS <- mxMatrix( type="Full", nrow=1, ncol=1,
free=TRUE, values=c(.3333), name="S")
# Matrix of observed data
obsFreq <- mxMatrix( type="Full", nrow=4, ncol=1,
values=c(211,104,39,148), name="ObservedFreqs")
matQ <- mxAlgebra( expression=1-P, name="Q")
matT <- mxAlgebra( expression=1-S, name="T")
# Algebra for predicted proportions
expFreq <- mxAlgebra( rbind ((P*P+2*P*Q)*T*T, (Q*Q)*(S*S+2*S*T),
(P*P+2*P*Q)*(S*S+2*S*T), (Q*Q)*(T*T)), name="ExpectedFreqs")
# Algebra for -2logLikelihood
m2ll <- mxAlgebra( expression=-(sum(log(ExpectedFreqs) * ObservedFreqs)),
name="NegativeLogLikelihood")
# User-defined objective
funAl <- mxFitFunctionAlgebra("NegativeLogLikelihood")
TwoLocusModel <- mxModel("TwoLocus",
matP, matS, matQ, matT, obsFreq, expFreq, m2ll, funAl)
TwoLocusFit<-mxRun(TwoLocusModel)
TwoLocusFit$matrices
TwoLocusFit$algebras
Results¶
The allele frequencies estimated by this script should be 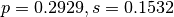 with negative log-likelihood of 646.972 units. Comparison of this model with the single locus one shows that although they have the same number of free parameters (the third allele frequency in the single locus model is constrained) the single locus model has much greater support. Investigation of the $ExpectedFreqs algebra in the two models helps to illustrate why.
with negative log-likelihood of 646.972 units. Comparison of this model with the single locus one shows that although they have the same number of free parameters (the third allele frequency in the single locus model is constrained) the single locus model has much greater support. Investigation of the $ExpectedFreqs algebra in the two models helps to illustrate why.