Genetic Epidemiology, Matrix Specification¶
Mx is probably most popular in the behavior genetics field, as it was conceived with genetic models in mind, which rely heavily on multiple groups. We introduce here an OpenMx script for the basic genetic model in genetic epidemiologic research, the ACE model. This model assumes that the variability in a phenotype, or observed variable, of interest can be explained by differences in genetic and environmental factors, with A representing additive genetic factors, C shared/common environmental factors and E unique/specific environmental factors (see Neale & Cardon 1992, for a detailed treatment). To estimate these three sources of variance, data have to be collected on relatives with different levels of genetic and environmental similarity to provide sufficient information to identify the parameters. One such design is the classical twin study, which compares the similarity of identical (monozygotic, MZ) and fraternal (dizygotic, DZ) twins to infer the role of A, C and E.
The example starts with the ACE model and includes one submodel, the AE model. It is available in the following file:
A parallel version of this example, using path specification of models rather than matrices, can be found here:
ACE Model: a Twin Analysis¶
Data¶
Let us assume you have collected data on a large sample of twin pairs for your phenotype of interest. For illustration purposes, we use Australian data on body mass index (BMI) which are saved in a text file ‘myTwinData.txt’. We use R to read the data into a data.frame and to create two subsets of the data for MZ females (mzData) and DZ females (dzData) respectively with the code below.
require(OpenMx)
#Prepare Data
data(myTwinData)
twinVars <- c( 'fam','age','zyg','part',
'wt1','wt2','ht1','ht2','htwt1','htwt2','bmi1','bmi2')
summary(myTwinData)
selVars <- c('bmi1','bmi2')
mzData <- as.matrix(subset(myTwinData, zyg==1, c(bmi1,bmi2)))
dzData <- as.matrix(subset(myTwinData, zyg==3, c(bmi1,bmi2)))
colMeans(mzData,na.rm=TRUE)
colMeans(dzData,na.rm=TRUE)
cov(mzData,use="complete")
cov(dzData,use="complete")
Model Specification¶
There are a variety of ways to set up the ACE model. The most commonly used approach in Mx is to specify three matrices for each of the three sources of variance. The matrix a represents the additive genetic path a, the c matrix is used for the shared environmental path c and the matrix e for the unique environmental path e. The expected variances and covariances between member of twin pairs are typically expressed in variance components (or the square of the path coefficients, i.e. 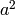 ,
, 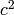 and
and 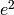 ). These quantities can be calculated using matrix algebra, by multiplying the a matrix by its transpose t(a), and are called A, C and E respectively. Note that the transpose is not strictly needed in the univariate case, but will allow easier transition to the multivariate case. We then use matrix algebra again to add the relevant matrices corresponding to the expectations for each of the statistics of the observed covariance matrix. The R functions ‘cbind’ and ‘rbind’ are used to concatenate the resulting matrices in the appropriate way. The expectations can be derived from the path diagrams for MZ and DZ twins.
). These quantities can be calculated using matrix algebra, by multiplying the a matrix by its transpose t(a), and are called A, C and E respectively. Note that the transpose is not strictly needed in the univariate case, but will allow easier transition to the multivariate case. We then use matrix algebra again to add the relevant matrices corresponding to the expectations for each of the statistics of the observed covariance matrix. The R functions ‘cbind’ and ‘rbind’ are used to concatenate the resulting matrices in the appropriate way. The expectations can be derived from the path diagrams for MZ and DZ twins.
Note that in R, lower and upper case names are distinguishable so we are using lower case letters for the matrices representing path coefficients a, c and e, rather than X, Y and Z that classic Mx users have become familiar with. We continue to use the same upper case letters for matrices representing variance components A, C and E, corresponding to additive genetic (co)variance, shared environmental (co)variance and unique environmental (co)variance respectively, calculated as the square of the path coefficients.
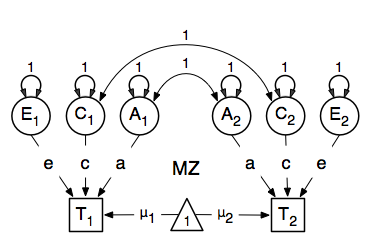
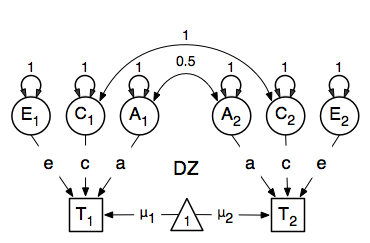
Let’s go through each of the matrices step by step. First, we start with the require(OpenMx) statement. We include the full code here. As MZ and DZ have to be evaluated together, the models for each will be arguments of a bigger model. Given the models for the MZ and the DZ group look rather similar, we start by specifying all the common elements in yet another model, called ACE which will then be evaluated together with the two submodels for each of the twin types, defined in separate mxModel commands, as they are all three arguments of the overall twinACE model, and will be saved together in the R object twinACEModel and thus be run together.
require(OpenMx)
twinACEModel <- mxModel("twinACE",
mxModel("ACE",
# Matrices a, c, and e to store a, c, and e path coefficients
mxMatrix(
type="Lower",
nrow=1,
ncol=1,
free=TRUE,
values=0.6,
labels="a11",
name="a"
),
mxMatrix(
type="Lower",
nrow=1,
ncol=1,
free=TRUE,
values=0.6,
labels="c11",
name="c"
),
mxMatrix(
type="Lower",
nrow=1,
ncol=1,
free=TRUE,
values=0.6,
labels="e11",
name="e"
),
# Matrices A, C, and E compute variance components
mxAlgebra(
expression=a %*% t(a),
name="A"
),
mxAlgebra(
expression=c %*% t(c),
name="C"
),
mxAlgebra(
expression=e %*% t(e),
name="E"
),
# Matrix & Algebra for expected means vector
mxMatrix(
type="Full",
nrow=1,
ncol=1,
free=TRUE,
values=20,
label="mean",
name="Mean"
),
mxAlgebra(
expression= cbind(Mean,Mean),
name="expMean"
),
# Algebra for expected variance/covariance matrix in MZ
mxAlgebra(
expression=rbind (cbind(A + C + E , A + C),
cbind(A + C , A + C + E)),
name="expCovMZ"
),
# Algebra for expected variance/covariance matrix in DZ
mxAlgebra(
expression=rbind (cbind(A + C + E , 0.5 %x% A + C),
cbind(0.5 %x% A + C , A + C + E)),
name="expCovDZ"
)
),
mxModel("MZ",
mxData(
observed=mzData,
type="raw"
),
mxFIMLObjective(
covariance="ACE.expCovMZ",
means="ACE.expMean",
dimnames=selVars
)
),
mxModel("DZ",
mxData(
observed=dzData,
type="raw"
),
mxFIMLObjective(
covariance="ACE.expCovDZ",
means="ACE.expMean",
dimnames=selVars
)
),
mxAlgebra(
expression=MZ.objective + DZ.objective,
name="minus2loglikelihood"
),
mxAlgebraObjective("minus2loglikelihood")
)
twinACEFit <- mxRun(twinACEModel)
They will all form arguments of the mxModel, specified as follows. Note that we left the comma’s at the end of the lines which are necessary when all the arguments are combined prior to running the model. Each line can be pasted into R, and then evaluated together once the whole model is specified.
#Fit ACE Model with RawData and Matrix-style Input
twinACEModel <- mxModel("twinACE",
mxModel("ACE",
Given the current example is univariate (in the sense that we analyze one variable, even though we have measured it in two members of twin pairs), the matrices for the paths a, c and e are all Full 1x1 matrices assigned the free status and given a 0.6 starting value.
# Matrices a, c, and e to store a, c, and e path coefficients
# additive genetic path
mxMatrix(
type="Full",
nrow=1,
ncol=1,
free=TRUE,
values=0.6,
label="a11",
name="a"
),
# shared environmental path
mxMatrix(
type="Full",
nrow=1,
ncol=1,
free=TRUE,
values=0.6,
label="c11",
name="c"
),
# specific environmental path
mxMatrix(
type="Full",
nrow=1,
ncol=1,
free=TRUE,
values=0.6,
label="e11",
name="e"
),
While the labels in these matrices are given lower case names, similar to the convention that paths have lower case names, the names for the variance component matrices, obtained from multiplying matrices with their transpose have upper case letters A, C and E which are distinct (as R is case-sensitive).
# Matrices A, C, and E compute variance components
# additive genetic variance
mxAlgebra(
expression=a * t(a),
name="A"
),
# shared environmental variance
mxAlgebra(
expression=c * t(c),
name="C"
),
# specific environmental variance
mxAlgebra(
expression=e * t(e),
name="E"
),
As the focus is on individual differences, the model for the means is typically simple. We can estimate each of the means, in each of the two groups (MZ & DZ) as free parameters. Alternatively, we can establish whether the means can be equated across order and zygosity by fitting submodels to the saturated model. In this case, we opted to use one ‘grand’ mean, obtained by assigning the same label to the elements of the matrix expMean by concatenating the Full 1x1 matrix Mean with one free element, labeled mean and given a start value of 20. The expMean matrix is then used in both the MZ and DZ model so that all four elements representing means are equated.
# Matrix & Algebra for expected means vector
mxMatrix(
type="Full",
nrow=1,
ncol=1,
free=TRUE,
values=20,
label="mean",
name="Mean"
),
mxAlgebra(
expression= cbind(Mean,Mean),
name="expMean"
),
Previous Mx users will likely be familiar with the look of the expected covariance matrices for MZ and DZ twin pairs. These 2x2 matrices are built by horizontal and vertical concatenation of the appropriate matrix expressions for the variance, the MZ or the DZ covariance. In R, concatenation of matrices is accomplished with the rbind and cbind functions. Thus to represent the matrices in expression below in R, we use the following code.
![\begin{eqnarray*}
covMZ = \left[ \begin{array}{c c} a^2+c^2+e^2 & a^2+c^2 \\
a^2+c^2 & a^2+c^2+e^2 \end{array} \right]
\end{eqnarray*}
\begin{eqnarray*}
covDZ = \left[ \begin{array}{c c} a^2+c^2+e^2 & .5a^2+c^2 \\
.5a^2+c^2 & a^2+c^2+e^2 \end{array} \right]
\end{eqnarray*}](_images/math/6b8000f33ac6e120ea38c10166a2e73dfa2e1054.png)
# Algebra for expected variance/covariance matrix in MZ
mxAlgebra(
expression=rbind (cbind(A + C + E , A + C),
cbind(A + C , A + C + E)),
name="expCovMZ"
),
# Algebra for expected variance/covariance matrix in DZ
mxAlgebra(
expression=rbind (cbind(A + C + E , 0.5 %x% A + C),
cbind(0.5 %x% A + C , A + C + E)),
name="expCovDZ"
)
),
As the expected covariance matrices are different for the two groups of twins, we specify two mxModel commands within the ‘twinACE’ mxModel command. They are given a name, and arguments for the data and the objective function to be used to optimize the model. We have set the model up for raw data, and thus will use the mxFIMLObjective function to evaluate it. For each model, the mxData command calls up the appropriate data, and provides a type, here raw, and the mxFIMLObjective command is given the names corresponding to the respective expected covariance matrices and mean vectors, specified above. Given the objects expCovMZ, expCovDZ and expMean were created in the mxModel named twinACE we need to use two-level names, starting with the model name separated from the object with a dot, i.e. twinACE.expCovMZ.
mxModel("MZ",
mxData(
observed=mzData,
type="raw"
),
mxFIMLObjective(
covariance="ACE.expCovMZ",
means="ACE.expMean",
dimnames=selVars
)
),
mxModel("DZ",
mxData(
observed=dzData,
type="raw"
),
mxFIMLObjective(
covariance="ACE.expCovDZ",
means="ACE.expMean",
dimnames=selVars
)
),
Finally, both models need to be evaluated simultaneously. We first generate the sum of the objective functions for the two groups, using mxAlgebra. We refer to the correct objective function (object named objective) by adding the name of the model to the two-level argument, i.e. MZ.objective. We then use that as argument of the mxAlgebraObjective command.
mxAlgebra(
expression=MZ.objective + DZ.objective,
name="minus2loglikelihood"
),
mxAlgebraObjective("minus2loglikelihood")
)
Model Fitting¶
We need to invoke the mxRun command to start the model evaluation and optimization. Detailed output will be available in the resulting object, which can be obtained by a print() statement.
#Run ACE model
twinACEFit <- mxRun(twinACEModel)
Often, however, one is interested in specific parts of the output. In the case of twin modeling, we typically will inspect the expected covariance matrices and mean vectors, the parameter estimates, and possibly some derived quantities, such as the standardized variance components, obtained by dividing each of the components by the total variance. Note in the code below that the mxEval command allows easy extraction of the values in the various matrices/algebras which form the first argument, with the model name as second argument. Once these values have been put in new objects, we can use and regular R expression to derive further quantities or organize them in a convenient format for including in tables. Note that helper functions could (and will likely) easily be written for standard models to produce ‘standard’ output.
MZc <- mxEval(ACE.expCovMZ, twinACEFit)
DZc <- mxEval(ACE.expCovDZ, twinACEFit)
M <- mxEval(ACE.expMean, twinACEFit)
A <- mxEval(ACE.A, twinACEFit)
C <- mxEval(ACE.C, twinACEFit)
E <- mxEval(ACE.E, twinACEFit)
V <- (A+C+E)
a2 <- A/V
c2 <- C/V
e2 <- E/V
ACEest <- rbind(cbind(A,C,E),cbind(a2,c2,e2))
LL_ACE <- mxEval(objective, twinACEFit)
Alternative Models: an AE Model¶
To evaluate the significance of each of the model parameters, nested submodels are fit in which these parameters are fixed to zero. If the likelihood ratio test between the two models is significant, the parameter that is dropped from the model significantly contributes to the phenotype in question. Here we show how we can fit the AE model as a submodel with a change in one mxMatrix command. First, we call up the previous ‘full’ model and save it as a new model twinAEModel. Next we re-specify the matrix c to be fixed to zero. We can run this model in the same way as before and generate similar summaries of the results.
#Run AE model
twinAEModel <- mxRename(twinACEModel, "twinAE")
# drop shared environmental path
twinAEModel$ACE.c <-
mxMatrix(
type="Full",
nrow=1,
ncol=1,
free=F,
values=0,
label="c11"
)
twinAEFit <- mxRun(twinAEModel)
MZc <- mxEval(ACE.expCovMZ, twinAEFit)
DZc <- mxEval(ACE.expCovDZ, twinAEFit)
A <- mxEval(ACE.A, twinAEFit)
C <- mxEval(ACE.C, twinAEFit)
E <- mxEval(ACE.E, twinAEFit)
V <- (A+C+E)
a2 <- A/V
c2 <- C/V
e2 <- E/V
AEest <- rbind(cbind(A,C,E),cbind(a2,c2,e2))
LL_AE <- mxEval(objective, twinAEFit)
We use a likelihood ratio test (or take the difference between -2 times the log-likelihoods of the two models) to determine the best fitting model, and print relevant output.
LRT_ACE_AE <- LL_AE-LL_ACE
#Print relevant output
ACEest
AEest
LRT_ACE_AE
Note that the OpenMx team is currently working on better alternatives for dropping parameters. These models may also be specified using paths instead of matrices, which allow for easier submodel specification. See Genetic Epidemiology, Path Specification for path specification of these models.